BC3203
Background
In this tutorial we will work with a gff (Genome Features Format) file containing the locations of all the features (genes, exons, introns etc) on the human genome.
The guide for this tutorial provides instructions on command-line (BASH) code that you will need to run in order to generate datasets required for further processing with R. This reflects the reality of many bioinformatic workflows. Often we will start by using command-line tools to cut datasets down to a manageable size, or perform resource intensive tasks. We will then export data to a form that can be read into R and used for visualisation or further analysis.
Setup
Start by running the setup code. This will download and unpack example data used for this guide, and for the corresponding exercises.
bash setup.sh
After running the setup code you should the following files and folder in your data folder;
| Name | Description |
|---|---|
GCF_000001405.39_GRCh38.p13_genomic.gff |
Human genome annotations |
Chromosomes.xlsx |
Human chromosome information |
You can view the expected outputs for all questions at https://bc3203.bioinformatics.guide/2024/tutorials/06/expected_outputs
Features in the Human Genome
Take a look at the NCBI RefSeq page for the Human Genome. This provides a summary of the genome sequence as well as its annotation (ie inferences about where the genes are). Tools are provided via this page for downloading various files associated with the genome and its annotation. There are quite a few different files associated with a Genome for an organism. There are two main types;
- Sequences in FASTA format. This includes;
- genome: the actual sequences for each chromosome and mitochondrion
- transcript: sequences for all the individual mRNA’s corresponding to genes
- protein: translations of all the mRNA sequences
- Annotations. These are the positions (start, end) and descriptions of all the features (chromosomes, genes, exons, introns, coding sequences etc) in the genome. The positions given refer to a given sequence entry in the genome file (eg a chromosome) and the number of bases along that sequence where the feature exists.
In this assignment we will focus on the annotation file in gff format. It’s just a simple tabular format. There are 9 fields separated by tab characters. These are effectively “columns” in a table of data. The fields (columns) we are most interested in for now are just the first 5;
| Field | Description |
|---|---|
| seqname | name of the chromosome or scaffold |
| source | name of the program that generated this feature, or the data source (database or project name) |
| feature | feature type name, e.g. Gene, Variation, Similarity |
| start | Start position of the feature, with sequence numbering starting at 1 |
| end | End position of the feature, with sequence numbering starting at 1 |
Use the command below to inspect the first few lines of the gtf file. This command uses head to obtain the first few lines and cut to restrict the output to columns 1-5
head data/GCF_000001405.39_GRCh38.p13_genomic.gff | cut -f1-5
Notice that the first 5 lines are not part of the table. They all start with a # and provide some overall metadata about the whole file. Next count the total number of lines in the file to get an appreciation for how large it is;
wc -l data/GCF_000001405.39_GRCh38.p13_genomic.gff
We would like to read the file into R to work with it but it is 3.8 Million lines long. It’s certainly possible to read a file this big into R, but it will take a long time and use a lot of memory. Next, we will use a tool called grep and another called awk to cut the file down to size so that we can work with only the relevant parts of it in R.
Unix Text Utilities
The unix text processing utilities, awk, sed and grep are incredibly useful tools for transforming biological data. By combining these three utilities with other standard uniq tools such as cat, sort, uniq and wc we can accomplish almost any kind of text processing on the command-line.
Rather than trying to learn all these tools in one go we will encounter them gradually and learn them in the context of tasks we want to accomplish.
Recall from the gff format definition that the third column denotes the feature type. What types of features are included in a gff file? We will use unix text utilities to find out.
grep
Let’s start by removing the comment lines from the start of the file. We can do this using grep. At it’s core, grep allows you to search for patterns of text in documents.
The most common usage of grep is like this
grep <pattern> file1
So, if we wanted to filter the Human gene annotations so that they only contained the comment lines we could use this command;
head data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep '^#'
Note that a comment is denoted by the symbol, # at the start of a line. The pattern used in the command above is a little more complex because it also includes the special character ^ which tells grep to look for the string at the start on the line only.
Now let’s try modifying the behaviour of grep. Try giving it the option -v as shown in the code below;
head data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#'
What’s happened here? Take a look at the manual entry for grep.
man grep
When viewing the man page you can use spacebar to scroll down, b to scroll up, and q to quit. You should be able to find an entry that explains the -v option. It says;
-v, --invert-match
Selected lines are those not matching any of the specified patterns.
So when we used -v it excluded all of the comment lines. This is very useful because we can now process the file reliably, knowing that every line will have exactly 9 tab delimited columns
Let’s try showing a few additional lines
head -n 20 data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#'
We are only interested in the third column for now as this describes the feature type. We can extract this using cut as follows;
head -n 20 data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | cut -f 3
sort and uniq
The sort command can be used to sort text. The default behaviour of sort is to sort alphabetically on the first column of text but it can be used to sort numerically on specific columns.
Try using sort to arrange the feature types in alphabetical order
head -n 20 data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | cut -f 3 | sort
The uniq command finds unique occurrences in text and removes duplicates. In order to work properly the input to uniq must first be sorted. A very common combination of tools is therefore to pipe sort to uniq, ie sort | uniq.
Let’s try it out on the genomic features
head -n 20 data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | cut -f 3 | sort | uniq
Now have a look at the manual page for uniq. You should find an option -c which is described as follows
-c Precede each output line with the count of the number of times the line occurred in the input, followed by a single space.
We can use this to modify our command so that it will also include a count of the number of times each feature occurs
head -n 20 data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | cut -f 3 | sort | uniq -c
And finally, we can add another sort command, this time to sort numerically by the number of occurrences, putting the features with the most occurrences at the top.
head -n 20 data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | cut -f 3 | sort | uniq -c | sort -n -r
So far we have worked with just a tiny fraction of the feature file. This is a good way to work when you are building up a command, figuring out how to get all the pieces in place. Now that we have things working let’s apply it to the entire file. We will also pipe our final output to head so that we see the top few, most abundant features in the file
cat data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | cut -f 3 | sort | uniq -c | sort -n -r | head
We can see that the most common genomic features are exons, CDS, mRNA and gene in that order. Notice that there are more mRNA entries than gene entries. This is because alternative splicing can give rise to multiple mRNA entries per gene.
Our goal is to use R tidyverse functions to explore the properties of gene entries in this file. We would therefore like to create a new version of this genome annotation file that only contains entries where the third column matches the keyword gene. To accomplish this, we will introduce a new tool, called awk.
AWK
The awk command processes text line by line. The awk programming language is used to specify actions to be performed on each line.
This illustrates the general usage of awk which is
awk 'program' file
Or alternatively, when used with a pipe
cat file | awk 'program'
Where the placeholder text, ‘program’ is a small program written in the awk language. In general awk programs are organised like this;
pattern { action }
Here’s how we could use awk to extract lines from the gff file where the third column is equal to “gene”.
head -n 100 data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | awk '$3=="gene"{print}'
In this case the awk program is $3=="gene"{print}. It consists of two parts. The pattern, $3=="gene" tests each line to see if the third column is equal to “gene”. In cases where the pattern finds a match the action will be executed. In this case the action is simply to print the entire line.
We are now in a position to generate a file for reading and manipulating in R. Run the following command to generate a new file cache/human_genes.gff
cat data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | awk '$3=="gene"{print}' > cache/human_genes.gff
STOP Answer Questions 1-3 in the exercises
Chromosomes in the Human Genome
Now let’s take a look at the first column of the Human genome gff file. This column encodes the seqid, a unique code that identifies the sequence on which the features exist. Firstly we want to know how many unique sequence ids there are in the file.
cat data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | cut -f 1 | sort | uniq | wc -l
Given that the human genome has only 22 autosomes, two sex chromosome (X,Y) and a mitochondrial sequence (MT) this is far more sequences than we expected. Many of the extra sequences in the Human genome are included in the file as a way to represent alternate haplotypes. This is important for representing human genetic variation and is something that you will not find in the genomes of most organisms. To keep things simple we would like to focus largely on the classical chromosomes and MT genome.
Using the same principles that we used to summarise the feature type column (column 3) we can summarise the sequence ID’s as follows;
cat data/GCF_000001405.39_GRCh38.p13_genomic.gff | grep -v '^#' | cut -f 1 | sort | uniq -c | sort -n -r | head -n 30
This command tells us the top 30 sequences in terms of numbers of gff entries. We can see that the top entries all have identifiers that look like NC_0000XX.YY where XX ranges from 0-24. These are most likely the autosomes and sex chromosomes.
The following code can be used to read a table with information on all of the Human chromosomes. Note that the entries in the RefSeq column correspond to the top entries in the summary we made above.
readxl::read_excel("data/Chromosomes.xlsx")
## # A tibble: 25 × 12
## Type Name RefSeq INSDC `Size (Mb)` `GC%` Protein rRNA tRNA `Other RNA`
## <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <chr> <chr> <chr>
## 1 Chr 1.0 NC_00000… CM00… 249. 42.3 11378 17.0 90.0 4605.0
## 2 Chr 2.0 NC_00000… CM00… 242. 40.3 8495 - 7.0 3850.0
## 3 Chr 3.0 NC_00000… CM00… 198. 39.7 7559 - 4.0 2849.0
## 4 Chr 4.0 NC_00000… CM00… 190. 38.3 4780 - 1.0 2258.0
## 5 Chr 5.0 NC_00000… CM00… 182. 39.5 4828 - 17.0 2280.0
## 6 Chr 6.0 NC_00000… CM00… 171. 39.6 5745 - 138.0 2583.0
## 7 Chr 7.0 NC_00000… CM00… 159. 40.7 5357 - 22.0 2480.0
## 8 Chr 8.0 NC_00000… CM00… 145. 40.2 4156 - 4.0 2025.0
## 9 Chr 9.0 NC_00000… CM00… 138. 42.3 4709 - 3.0 2289.0
## 10 Chr 10.0 NC_00001… CM00… 134. 41.6 5531 - 3.0 2234.0
## # ℹ 15 more rows
## # ℹ 2 more variables: Gene <dbl>, Pseudogene <chr>
Correct ordering in plots
Later in this tutorial we will create plots in which the data is grouped by chromosome. We want chromosomes to appear in their natural order, ie 1-22, X, Y, MT. This isn’t as easy as it might seem. The following plot illustrates the problem;
readxl::read_excel("data/Chromosomes.xlsx") %>%
ggplot(aes(x=`Size (Mb)`,y=Name)) +
geom_col()
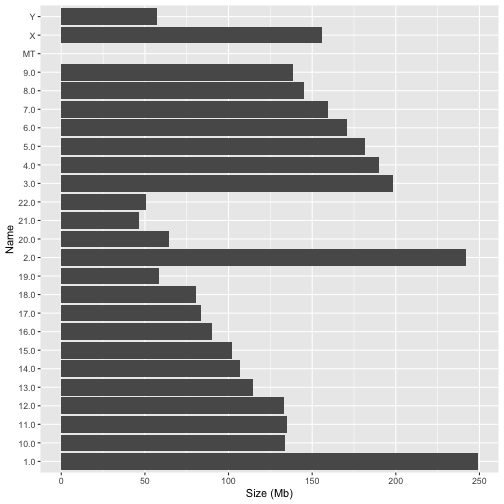
Notice how ggplot reorders the chromosomes in an unnatural way. This is because the Name field is of type character and ggplot will impose alphabetical ordering on it.
A good way to address this issue is to create an extra column that encodes the order with a numerical variable. For the chromosome table above this is easy because the chromosomes are already provided in the correct order. We can therefore add a column with the row number as follows;
readxl::read_excel("data/Chromosomes.xlsx") %>%
mutate(chr_order = row_number())
## # A tibble: 25 × 13
## Type Name RefSeq INSDC `Size (Mb)` `GC%` Protein rRNA tRNA `Other RNA`
## <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <chr> <chr> <chr>
## 1 Chr 1.0 NC_00000… CM00… 249. 42.3 11378 17.0 90.0 4605.0
## 2 Chr 2.0 NC_00000… CM00… 242. 40.3 8495 - 7.0 3850.0
## 3 Chr 3.0 NC_00000… CM00… 198. 39.7 7559 - 4.0 2849.0
## 4 Chr 4.0 NC_00000… CM00… 190. 38.3 4780 - 1.0 2258.0
## 5 Chr 5.0 NC_00000… CM00… 182. 39.5 4828 - 17.0 2280.0
## 6 Chr 6.0 NC_00000… CM00… 171. 39.6 5745 - 138.0 2583.0
## 7 Chr 7.0 NC_00000… CM00… 159. 40.7 5357 - 22.0 2480.0
## 8 Chr 8.0 NC_00000… CM00… 145. 40.2 4156 - 4.0 2025.0
## 9 Chr 9.0 NC_00000… CM00… 138. 42.3 4709 - 3.0 2289.0
## 10 Chr 10.0 NC_00001… CM00… 134. 41.6 5531 - 3.0 2234.0
## # ℹ 15 more rows
## # ℹ 3 more variables: Gene <dbl>, Pseudogene <chr>, chr_order <int>
We can now adjust our plotting code, making use of the function reorder to tell ggplot a column to use for the ordering
readxl::read_excel("data/Chromosomes.xlsx") %>%
mutate(chr_order = row_number()) %>%
ggplot(aes(x=`Size (Mb)`,y=reorder(Name,chr_order))) +
geom_col()
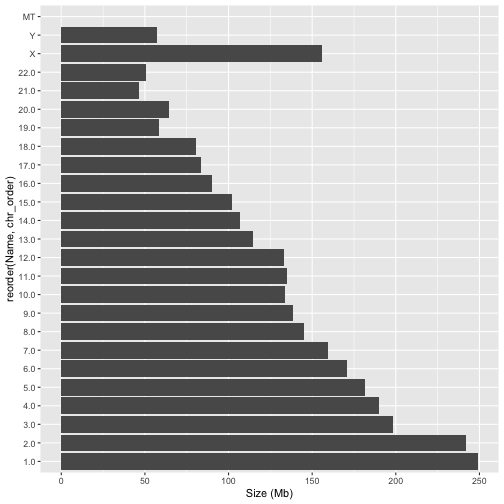
STOP Answer Questions 4-8 in the exercises
Regular Expressions
Earlier in this guide we extracted human gene entries into a file called human_genes.gff. In doing this we used grep, which is a command-line tool that searches for patterns in text. grep stands for Global Regular-Expression and Print. The regular expression part of the name comes from the fact that grep uses a specialised syntax called regular expressions to specify the patterns for searching. Our first encounter with regular expressions was the expresion
^#
This uses the special character ^ to indicate that the match should start from the beginning of the text. Then the # indicates a character to match.
Aside from ^, there are many other special characters used in regular expressions. Some of the most common are;
| Operator | Description |
|---|---|
^ |
Match the start of the text |
$ |
Match the end of the text |
. |
Match any character |
[0-9] |
Match a numeric digit between 0 and 9 |
\w |
Match a word character |
* |
Match an character group any number of times |
+ |
Match an character group one or more times |
[^<char>] |
Where <char> stands for a character group. Match anything except for <char> |
The full syntax of regular expressions is fairly complex, and is made extra difficult by the fact that different regex engines sometimes work differently. Don’t let this put you off though. If you learn just a few basic features of regular expressions you can accomplish a great deal.
As an example for working with regular expressions let’s take the first 10 entries in the human_genes.gff file that we created earlier. We can do this in R as follows;
gff_cols <- c("seqid","source","type","start","end","score","strand","phase","attributes")
gff10 <- read_tsv("cache/human_genes.gff",col_names = gff_cols,comment = "#", n_max = 10)
Inspect the 9th column of this dataset (called attributes) It has entries that look like this;
"ID=gene-MIR6859-1;Dbxref=GeneID:102466751,HGNC:HGNC:50039,miRBase:MI0022705;Name=MIR6859-1;description=microRNA 6859-1;gbkey=Gene;gene=MIR6859-1;gene_biotype=miRNA;gene_synonym=hsa-mir-6859-1"
Notice how all the entries in the attributes column are structured into key-value pairs separated by a semicolon “;”. For example;
ID=gene-MIR6859-1
Dbxref=GeneID:102466751,HGNC:HGNC:50039
Now let’s try some simple regular expression matching on this field. If we want to filter for rows that match a regex we can use the str_detect function in conjunction with filter from dplyr. For example, this code will extract only rows corresponding to genes that encode a particular type of transcript called a microRNA (miRNA);
gff10 %>% filter(str_detect(attributes,"gene_biotype=miRNA"))
## # A tibble: 3 × 9
## seqid source type start end score strand phase attributes
## <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr> <chr>
## 1 NC_000001.11 BestRefSeq gene 17369 17436 . - . ID=gene-MIR685…
## 2 NC_000001.11 BestRefSeq gene 30366 30503 . + . ID=gene-MIR130…
## 3 NC_000001.11 BestRefSeq gene 187891 187958 . - . ID=gene-MIR685…
Micro RNA’s are a particularly interesting type of gene product. They don’t encode proteins, but are involved in regulating expression of other genes. If we want to extract the entries for protein coding genes we could use;
gff10 %>% filter(str_detect(attributes,"gene_biotype=protein_coding"))
## # A tibble: 2 × 9
## seqid source type start end score strand phase attributes
## <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr> <chr>
## 1 NC_000001.11 BestRefSeq gene 65419 71585 . + . ID=gene-OR4F5;…
## 2 NC_000001.11 Gnomon gene 350706 476822 . - . ID=gene-LOC112…
Now what if our goal was not just to filter rows, but to extract the value of gene_biotype in every row. This can be accomplished using another aspect of regular expressions, called a capture group. If we want to capture a portion of text that matches a pattern we can simply enclose the pattern in brackets (<pattern>). To actually perform the extraction we also use a different dplyr function called extract. Here is an example;
gff10 %>% extract(attributes, "biotype","gene_biotype=([^;]*)")
## # A tibble: 10 × 9
## seqid source type start end score strand phase biotype
## <chr> <chr> <chr> <dbl> <dbl> <chr> <chr> <chr> <chr>
## 1 NC_000001.11 BestRefSeq gene 17369 17436 . - . miRNA
## 2 NC_000001.11 Gnomon gene 29926 31295 . + . lncRNA
## 3 NC_000001.11 BestRefSeq gene 30366 30503 . + . miRNA
## 4 NC_000001.11 BestRefSeq gene 34611 36081 . - . lncRNA
## 5 NC_000001.11 BestRefSeq gene 65419 71585 . + . protein_coding
## 6 NC_000001.11 Gnomon gene 89006 267354 . - . lncRNA
## 7 NC_000001.11 BestRefSeq gene 134773 140566 . - . lncRNA
## 8 NC_000001.11 BestRefSeq gene 187891 187958 . - . miRNA
## 9 NC_000001.11 Gnomon gene 200442 203983 . + . lncRNA
## 10 NC_000001.11 Gnomon gene 350706 476822 . - . protein_coding
There is a fair bit going on in the regular expression here. Let’s start with the part inside the capture group, which is [^;]*. This part in square brackets matches any character except for a semicolon. This is useful because the semicolon delimits each entry in the attributes string. The * after the square brackets indicates that any number of characters should be matched (except for a ;). The text that comes before the capture group effectively anchors the match by searching for text that matches gene_biotype=. This ensures that the capture group will start after this text and continue until the first semicolon.
STOP Answer Questions 9-10 in the exercises